UMI-based variant calling
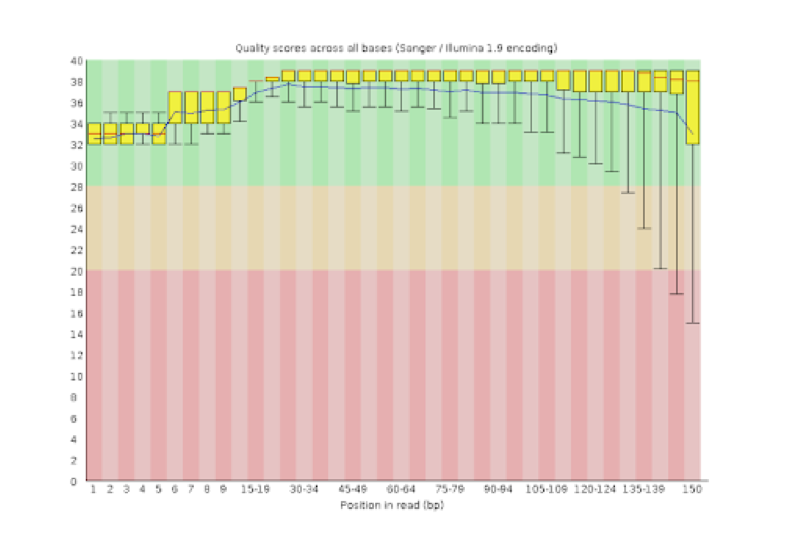
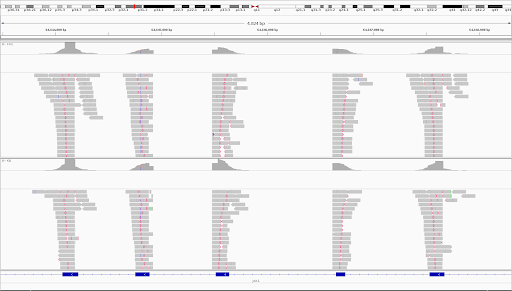
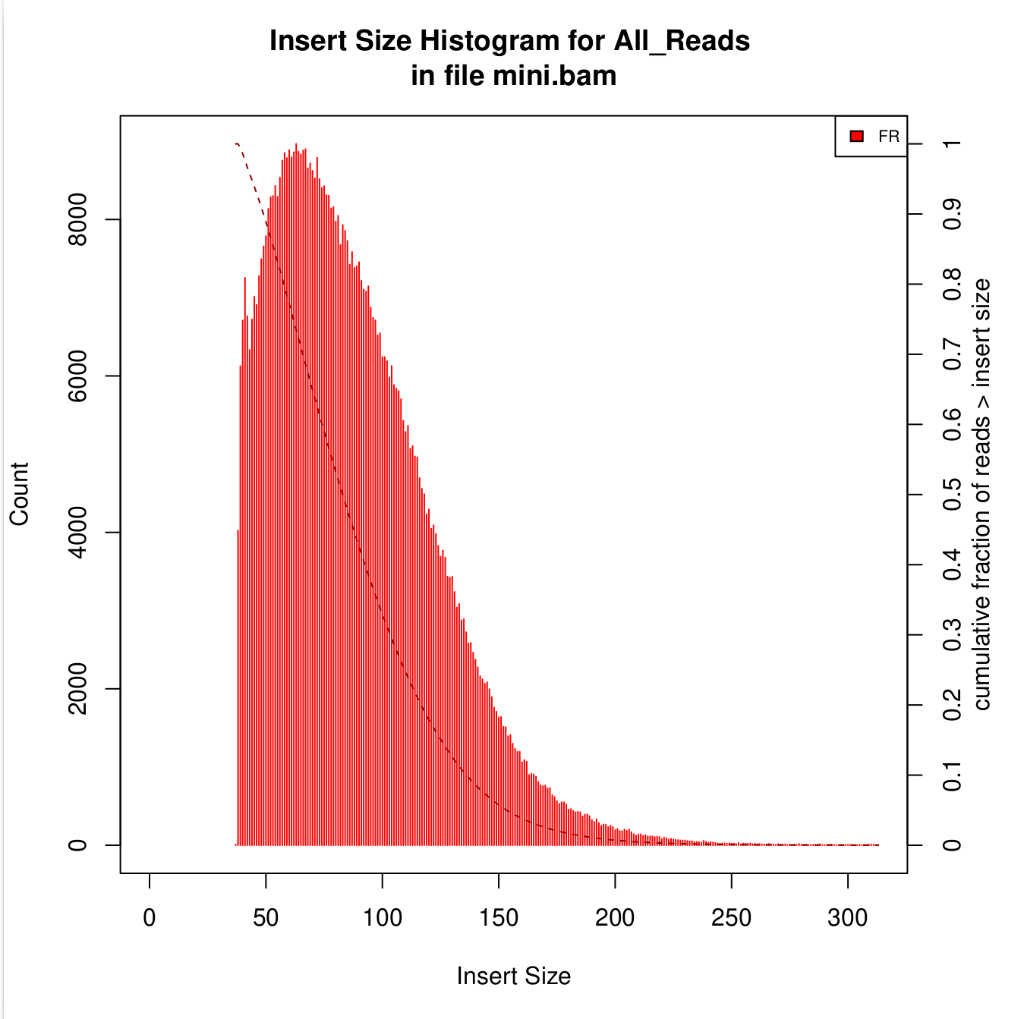
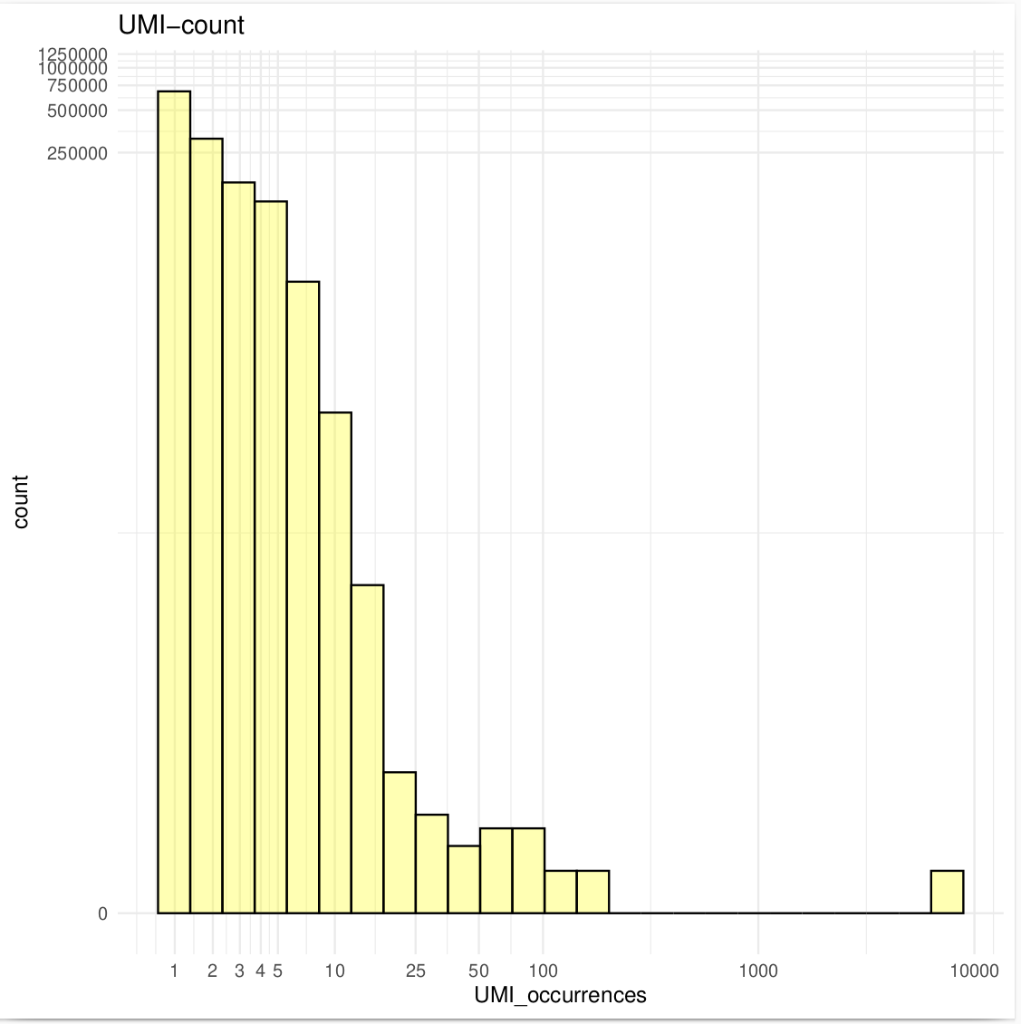
Unique molecular identifiers (UMIs) are a type of molecular barcoding that provides error correction and increased accuracy on Next Generation Sequencing (NGS) data. UMIs allow for the identification of PCR duplicates and the correction of sequencing errors, which are common sources of false positives.
Sequencing accuracy using UMI-based reads significantly reduces false positives and increases variant calling sensitivity, allowing to confidently call variants with a frequency below 1%.
 Book a Demo
Book a Demo Run Your Analysis
Run Your Analysis